作者:傅兴 R语言中文社区专栏作者
个人公众号:Rapp
从2015年开始,全世界掀起了一股“精准医疗”的狂潮,癌症基因组测序迅速成为生物医学行业关注的焦点。其实,早在“精准医疗”口号响起之前,美国人已经做了很多癌症基因组测序的工作,鼎鼎大名的就属TCGA(The Cancer Genome Atlas)。
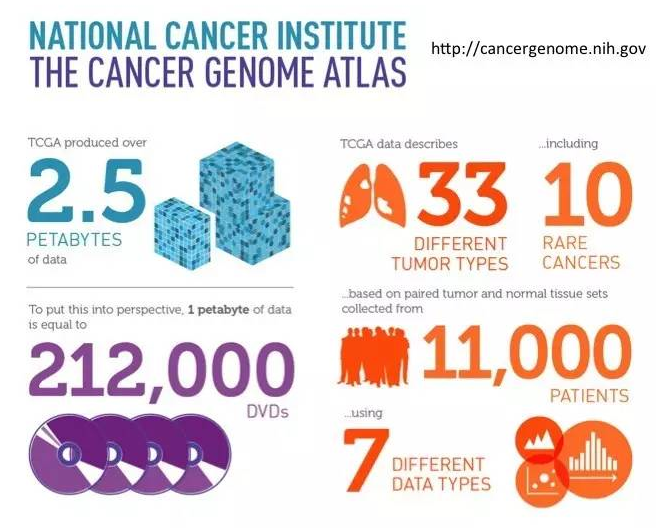
TCGA上的数据量庞大,数据种类丰富,分析方法复杂,并不是所有人都能轻松下载、管理和分析这些数据。对于大部分研究人员来说,从如此海量的原始测序数据开始分析是不可行也是不必要的。实际上,我们可以下载经过预处理后的数据(pre-processed data),不仅数据量会小很多,分析起来也更快、更可靠。Broad institute开发的Firehose就能够提供这样的数据。

有TCGA的大数据做基础,也有Firehose为我们做好前期的处理工作,现在就缺一个“搜索引擎”。今天就给大家介绍一个这样的利器:RTCGAToolbox

RTCGAToolbox是Bioconductor上的一个软件包,它的作用就是查询、下载和组织TCGA Firehose的数据,还提供一些简单的数据分析和可视化工具。除此之外,下载好的数据也可以很方便的导入到Bioconductor的其他分析流程中。对于R用户来说,所有的TCGA数据分析工作(从数据下载一直到可视化图表)都可在一个pipeline中完成,能够极大地提高工作效率。RTCGAToolbox的基本工作流程如下:
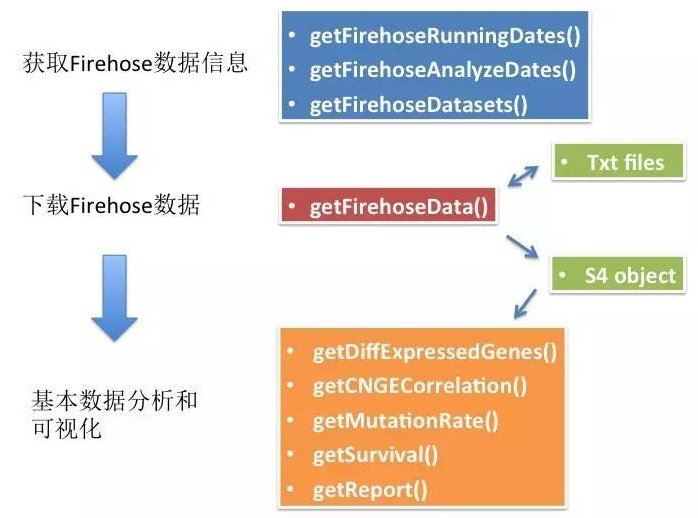
下面就简单介绍一下如何用RTCGAToolbox分析TCGA数据:
1. 首先,我们可以用getFirehoseDatasets函数查看Firehose上所有的肿瘤类型,从中选择我们感兴趣的数据。Firehose的数据有很多版本,每个版本都是用分析日期做为标记,所以在下载之前,可以使用getFirehoseRunningDates和getFirehoseAnalyzeDates函数来查询一下有哪些可供下载的版本。一般我们都会选择最新的版本,当我们要重复别人的结果时,最好选择一样的版本。
2. 接下来,我们要选择下载什么类型的数据(临床数据,基因表达,CNV,DNA甲基化等等),RTCGAToolbox目前支持绝大部分Firehose的数据:
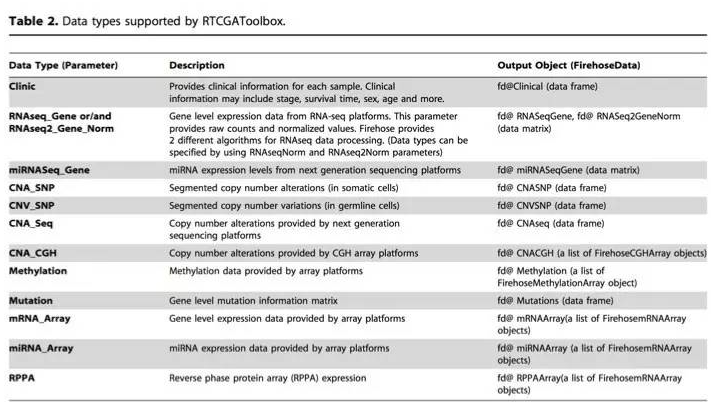
3. 当确定了上述所有的信息后,只需要把它们作为getFirehoseData函数的参数就可以轻松开始下载你所需要的数据了。
4. 下载后的数据会以文本文件的形式存放在当前工作目录下,同时,函数会返回一个S4的对象,该对象包含了所有已经下载的数据,后续的分析只需要处理这一个对象就可以了,非常方便。当你下次再执行getFirehoseData的时候,如果当前工作目录下的文件还在,它就直接读入,不会再去重新下载,考虑的非常周到。
RTCGAToolbox提供了5个基本的数据分析工具:
1. 差异表达分析(比较肿瘤和正常组织的基因表达量),根据不同的平台(RNA-Seq或Microarray),自动选择适合的工具
2. 拷贝数和基因表达量的相关性分析
3. 基因突变率分析
4. 生存分析
5. 可视化报告
下面是RTCGAToolbox输出的一些图:
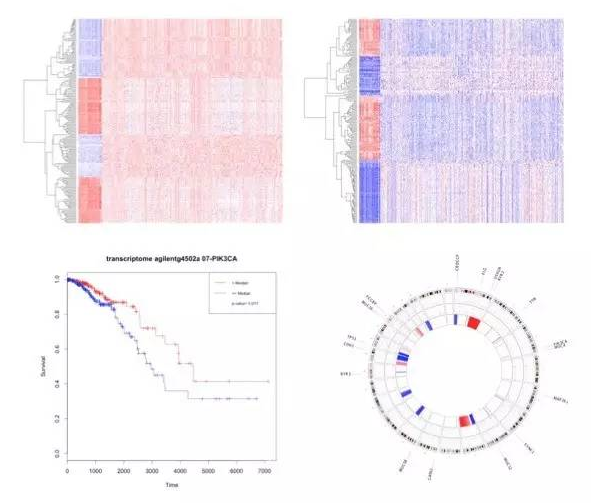
其实这5个工具是远远不够的(而且图画的也很难看),但是不用担心,Bioconductor环境下有非常多的统计分析和可视化工具供你选择。
我推荐RTCGAToolbox的原因有2点:
1. 通过一个函数自动完成所有数据下载的工作(包括下载,解压,读入文件,删除压缩文件),极为方便
2. 读入的TCGA数据被自动封装在一个S4的对象中,我们可以通过各种接口来轻松的访问它内部的数据,一个有条理的数据组织结构可以大大提高程序的可读性和可维护性
对R的面向对象编程不熟悉的同学可以继续关注Rapp公众号的生信训练营,我将会介绍更多的Bioconductor包,在这个过程中,大家会慢慢习惯用面向对象的思维方式来分析数据。
今天的介绍结束了,但是训练营才刚刚开始,赶快动手操作一下RTCGAToolbox吧!

